 Download Protocol PDF
Download Protocol PDFThe following table will help you to plan and integrate the different experimental methods.
| Experiment | Day |
Time |
Activity | |
| I. Collect, Document, and Identify Specimens | -1 |
varies |
Lab: | Collect tissue or processed material |
| II. Isolate DNA from Plant, Fungal, or Animal Samples | 1 |
30–60 min |
Pre-lab: | Aliquot* distilled water or TE, lysis solution,
wash buffer, and silica resin (silica protocol);
hole punch Whatman No. 1 chromatography
paper (rapid protocol) Set up student stations |
30/80 min |
Lab: | Isolate DNA (rapid/silica) | ||
| III. Amplify DNA by PCR | 2 |
15 min |
Pre-lab: | Prepare and aliquot* primer mix
Set up student stations |
10 min |
Lab: | Set up PCR reactions | ||
70–150 min |
Post-Lab: | Amplify DNA in thermal cycler | ||
| IV. Analyze PCR Products by Gel Electrophoresis | 3 |
30 min |
Pre-lab: | Dilute TBE electrophoresis buffer
Prepare agarose gel solution Set up student stations |
30 min |
Lab: | Cast gels | ||
| 45+ min |
Load DNA samples into gels
Electrophorese samples Photograph gels |
*Suggested volumes account for pipetting error and include about 20% additional reagent per tube.
- Collecting Samples
- Isolating DNA
- Primer Strategy and Design
- Barcode Primer Sequences
- Ordering Primers
- Ready-to-Go PCR Beads
- NEB Taq 2X Master Mix
- Primer/Loading Dye Mix
- Alternative PCR Protocol for NEB Taq 2X Master Mix
- Centrifuging PCR Tubes
- Thermal Cycling
- Troubleshooting Guide for Failed Reactions
- Agarose (2%)
- Gel Electrophoresis
- DNA Sequencing
- Using Genewiz DNA Sequencing Services
Brainstorm in class to identify one or more organized campaigns that students can be involved with. Students may select samples of their own choosing; this can be done as a homework assignment or in class if the season permits. Alternatively, teachers can provide samples.
Obtain permission to collect on private property, parks, or nature preserves.
One application of DNA barcoding is to survey species from a particular location or habitat. Since accounting for every plant and animal in a habitat is usually impossible, samples are collected to generally represent the habitat. A common sampling unit is a quadrat, a 1-meter square frame that is laid over the ground and from which each different plant and animal is collected for barcoding. Quadrats make it possible to compare samples from different locations or habitats. Nets are useful for collecting flying insects or swimming invertebrates. A sample of freshwater or marine organisms can be strained from a defined amount of water.
Plants and Fungi
Avoid collecting woody parts, which are difficult to break up, and starchy storage tissue, which includes metabolites that may interfere with PCR. If fresh green plants are not available, DNA is readily isolated from frozen or dried material. Students may also bring in items from the grocery store. Fresh produce works well, and many processed foods containing plant material will also work. It is difficult to isolate DNA from fatty or oily foods, such as peanut butter.
For fungi, obtain fruit bodies (such as mushrooms) when possible. Avoid contamination by other fungi, such as moldy mushrooms or multiple species growing together. Fresh samples from soft mushrooms work well for DNA isolation, while dried samples and hard fungi give variable results. Fungal fruiting is weather and climate dependent, so their abundance will vary, although both fresh and dried mushrooms are readily available from stores.
Animals
Insects offer great opportunities for barcoding. A kill jar is a simple and humane way to collect and kill insects. Make a kill jar from a wide-mouth plastic jar with tight fitting lid. Cut enough discs of paper towel to make a ½ inch stack in the bottom of the jar, then soak the toweling with acetone (nail polish remover). Keep the jar tightly capped, away from flames. Alternatively, kill insects by placing them in the freezer for at least one hour. Larger animals may be safely sampled, and without injury, by isolating DNA from hair, feathers, or dung. Hair roots (follicles) and flesh scraped from the base of the feather shaft are reliable sources. Fresh meats and fish from the grocery store are good sources of DNA for barcoding. Many processed foods, and food scraps obtained at no cost, are good sources of DNA.
Refer to table below to determine the appropriate isolation method and PCR primers for different sample types.
| Sample Type | Isolation Method $= Cost per sample, % = PCR success rates (for first choice primers), N/A: not applicable |
Recommended Primers for PCR |
DNA Extraction and Primer Tips | ||||
| IIa. Rapid DNA Isolation $ |
IIb. Silica DNA Isolation $$ |
IIc. Chelex DNA Isolation $ |
IId. QIAGEN DNeasy Blood and Tissue Kit
$$$ |
IIe. QIAGEN DNeasy
Plant Kit $$$ |
The first primer listed is recommended. Use alternate primer(s) if PCR fails. | Small sample sizes are required (approximately the size of a grain of rice). 3-mm tissue punch devices, such as surgical biopsy punches, may help to standardize sample preparation. Commercial kits are recommended for difficult (like highly processed, dry, and fatty) samples. | |
| Plants (Terrestrial) | 85% | 85% | 85% | N/A | 85% | 1: rbcL 2: matK 3: plant ITS |
Soak dry samples in water prior to DNA isolation. DNA extraction is more difficult from needles, seeds, bark, roots, waxy leaves, and cones. rbcL and matK may not resolve the sample to the species level. Plant ITS is an alternative but fewer reference sequences exist in sequence databases. |
| Plants (Aquatic) | 50% | 50% | TBD | N/A | 70% | 1: rbcL 2: matK 3: plant ITS |
rbcL and matK may not resolve the sample to the species level. Plant ITS is an alternative but fewer reference sequences exist in sequence databases. |
| Plants (Algae-specific) | 30% | 40% | TBD | N/A | 80% | 1: rbcL 2: tufA 3: plant ITS |
These primers are recommended for macroscopic green algae. |
| Invertebrates (Terrestrial) | 60% | 70% | Ant tested 70% | 80% | N/A | 1: Invertebrate COI |
For larger organisms, remove a small portion (example: abdomen of an ant, leg of a mosquito or spider). |
| Invertebrates (Freshwater) | 30% | 60% | TBD | 70% | N/A | 1: Invertebrate COI |
|
| Invertebrates (Marine) | 30% | 40% | TBD | 70% | N/A | 1: Invertebrate COI |
The rapid and silica isolation methods do not work across a broad spectrum of marine invertebrates; the Qiagen DNeasy Blood and Tissue kit is recommended. |
| Vertebrates (Fish) | 35% | 70% | TBD | 80% | N/A | Vertebrate (fish) cocktail COI | |
| Vertebrates (Non-Fish) | 60% | 70% | TBD | 80% | N/A | Vertebrate (non-fish) cocktail COI | |
| Fungi | 70% | 70% | TBD | N/A | 80% | Fungi ITS | |
| Fungi (lichen-specific) | 70% | 70% | TBD | N/A | 90% | Fungi (lichen-specific) ITS | Pre-wash samples with 100% acetone to remove lipids and polyphenol contaminants and let them dry for several hours before DNA isolation. |
Part IIa works well for plant and terrestrial invertebrates, with an affordable and very rapid isolation method using Whatman No. 1 chromatography paper.
Part IIb works well for plant, fungal, or animal samples and uses an affordable silica resin DNA isolation method.
Parts IId and IIe work well for animal, plant, or fungal samples, but use more expensive reagents from Qiagen® DNeasy Blood and Tissue Kit catalogue number 69506 (250 preps) or Qiagen® DNeasy Plant Kit, catalogue number 69106 (250 preps). These are recommended for difficult samples or for samples for which IIa or IIb fail to give DNA that amplifies.
10% Chelex Solution
Makes 50 mL.
Store at 4°C.
- Dissolve 5 g of Chelex 100 Resin in 35 mL of 50 mM Tris-HL buffer.
- Using a pH meter, adjust the pH to 11 using 4N NaOH.
- Add deionized or distilled water to make a total volume of 50 mL of solution.
Note: Before pipetting each aliquot, shake the stock tube to resuspend the Chelex resin, as it settles quickly.
Ethylenediaminetetraacetic Acid (EDTA) (0.5 M, pH 8.0)
Makes 100 mL.
Store at room temperature (indefinitely).
- Add 18.6 g of EDTA (disodium salt dihydrate, MW 372.24) to 80 mL of deionized or distilled water.
- Adjust the pH by slowly adding ~2.2 g of sodium hydroxide (NaOH) pellets (MW 40.00); monitor with a pH meter or strips of pH paper. (If neither is available, adding 2.2 g of NaOH pellets will make a solution of ~pH 8.0.)
- Mix vigorously with a magnetic stirrer or by hand.
- Add deionized or distilled water to make a total volume of 100 mL of solution.
- Make sure that the bottle cap is loose and autoclave for 15 min at 121° C.
- After autoclaving, cool the solution to room temperature and tighten the lid for storage.
Notes: Use only the disodium salt of EDTA. EDTA will only dissolve after the pH has reached 8.0 or higher.
6 M Guanidine Hydrochloride Solution
Makes 100 mL.
Store at room temperature (for 6 months).
- Dissolve 57.32 g of guanidine hydrochloride (m.w. = 95.53) in 50 mL of deonized or distilled water.
- Add deionized or distilled water to make a total volume of 100 mL of solution.
Paper Discs
Whatman No.1 chromatography paper (Carolina Biological Item # 689110)
3-mm hole punch
100% EtOH
- Clean a 3-mm hole punch with 100% EtOH, allow to dry. It is recommended that this hole punch is dedicated solely to creating the discs.
- Use hole punch to punch discs (1 per sample) from the Whatman No.1 chromatography paper.
- Store the discs in a sterile container, such as a 1.5-mL microcentrifuge tube.
Silica Resin Solution
Makes 50 mL.
Store at 4°C.
- Dissolve 25 g of silicon dioxide (m.w. = 60.08) in 35 mL of deionized or distilled water.
- Add deionized or distilled water to make a total volume of 50 mL of solution.
Note: The silica resin must be rinsed with 50 mL distilled water 3-4 times by centrifugation prior to bringing the total volume to 50 mL.
Sodium Chloride (NaCl) (5 M)
Makes 500 mL.
Store at room temperature (indefinitely).
- Dissolve 146.1 g of NaCl (MW 58.44) in 250 mL of deionized or distilled water.
- Add deionized or distilled water to make a total volume of 500 mL of solution.
Tris/EDTA (TE) Buffer
Makes 100 mL.
Store at room temperature (indefinitely).
- In a 200-mL beaker mix the following:
- 99 mL of deionized or distilled water
- 1 mL of 1 M Tris pH 8.0
- 200 µL of 0.5 M EDTA
- Mix well.
Tris-HCl (1 M, pH 8.0 and 8.3)
Makes 100 mL.
Store at room temperature (indefinitely).
- Dissolve 12.1 g of Tris base (MW 121.10) in 70 mL of deionized or distilled water.
- Adjust the pH by slowly adding concentrated hydrochloric acid (HCl) for the desired pH listed below.
- pH 8.0: 5.0 mL
- pH 8.3: 4.5 mL
- Monitor with a pH meter or strips of pH paper. (If neither is available, adding the volumes of concentrated HCl listed here will yield a solution with approximately the desired pH.)
- Add deionized or distilled water to make a total volume of 100 mL of solution.
- Make sure that the bottle cap is loose and autoclave for 15 min at 121°C.
- After autoclaving, cool the solution to room temperature and tighten the lid for storage.
Notes: A yellow-colored solution indicates poor-quality Tris. If your solution is yellow, discard it and obtain a Tris solution from a different source. The pH of Tris solutions is temperature dependent, so make sure to measure the pH at room temperature. Many types of electrodes do not accurately measure the pH of Tris solutions; check with the manufacturer to obtain a suitable one.
Wash Buffer
Makes 500 mL.
Store at -20°C (indefinitely).
- Combine the following:
- Deionized or distilled water, 234 mL
- 1 M Tris (pH 7.4), 10 mL
- 5 M NaCl, 5 mL
- 0.5 M EDTA, 1 mL
- 100% Ethanol, 250 mL
- Mix thoroughly.
DNA barcoding relies on finding a universal chromosome location (locus) that has retained enough sequence conservation through evolutionary history that it can be identified in many organisms, but that also has enough sequence diversity to differentiate organisms to at least the family level. Regions of the chloroplast rbcL gene, mitochondrial COI gene, and nuclear ITS region generally fulfill these requirements.
Primers are designed to target conserved sequences that flank the variable barcode regions. However, even the conserved flanking regions have accumulated enough sequence differences over evolutionary time that it is impossible to identify universal primer sets that will work across all taxonomic groups of plants and animals. Thus, barcode primers often need to accommodate sequence variation, or degeneracy, at one or several nucleotide positions.
The degeneracy problem is often solved when the oligonucleotide primers are synthesized. Traditionally, a mixture of primers is synthesized – each having a different nucleotide in any of the variable positions. However, synthetic nucleotides are now available that pair with multiple nucleotides and can be incorporated at variable positions in a single primer.
The table below shows the letter abbreviation given for degenerate nucleotides. For example, a primer with the sequence "ATCCR" contains both ATCCA and ATCCG.
| W = A or T S = G or C M = A or C K = G or T R = A or G Y = C or T |
B = C or G or T D = A or G or T H = A or C or T V = A or C or G N = A or C or G or T |
Even degenerate primers cannot ensure amplification in taxonomic groups in which all or part of a particular primer sequence is deleted. Thus, broad surveys of unknown plants or animals typically employ multiple primer sets against slightly different flanking regions, which are combined in a multiplex PCR reaction.
The rbcL primer set used in this laboratory will work well for most green plants. We suggest starting with one of three COI primer sets for animals: one for fish, one for vertebrates, and one for other invertebrates (DMI). For fungal species, use the ITS primer set, which has the highest chance of success for identifying a broad range of fungi. These primer sets will not uniformly work across all groups. If the primers in this laboratory do not work with a group of organisms you are studying, consult the primer list (http://www.boldsystems.org/index.php/Public_Primer_PrimerSearch) at the Barcode of Life Online Database web site for alternatives.
Primers |
Primer Sequence |
||
PLANT PRIMER SETS For use with plants Kress WJ, Erickson DL (2007) A Two-Locus Global DNA Barcode for Land Plants: The Coding rbcL Gene Complements the Non-Coding trnH-psbA Spacer Region. PLoS ONE 2(6) | |||
| rbcLaF | 5’- TGTAAAACGACGGCCAGTATGTCACCACAAACAGAGACTAAAGC-3’ | ||
| rbcLa rev | 5’- CAGGAAACAGCTATGACGTAAAATCAAGTCCACCRCG-3’ | ||
| matk-3F | 5’-TGTAAAACGACGGCCAGTCGTACAGTACTTTTGTGTTTACGAG-3’ | ||
| matk-1R | 5’-CAGGAAACAGCTATGACACCCAGTCCATCTGGAAATCTTGGTTC-3’ | ||
| nrITS2-S2F | 5’-TGTAAAACGACGGCCAGTATGCGATACTTGGTGTGAAT-3’ | ||
| nrITS2-S3R | 5’-CAGGAAACAGCTATGACGACGCTTCTCCAGACTACAAT-3’ | ||
| tufA_F | 5’-TGTAAAACGACGGCCAGTTGAAACAGAAMAWCGTCATTATGC-3’ | ||
| tufA_R | 5’-CAGGAAACAGCTATGACCCTTCNCGAATMGCRAAWCGC-3’ | ||
|
VERTEBRATE (FISH) PRIMER COCKTAIL For use with fish and some mammals Ivanova et. al. (2007). Universal primer cocktails for fish DNA barcoding. Molecular Ecology Notes 7, 544-548 | |||
| VF2_t1 | 5’-TGTAAAACGACGGCCAGTCAACCAACCACAAAGACATTGGCAC-3’ | ||
| FishF2_t1 | 5'’'- TGTAAAACGACGGCCAGTCGACTAATCATAAAGATATCGGCAC-3’ | ||
| FishR2_t1 | 5'’'- CAGGAAACAGCTATGACACTTCAGGGTGACCGAAGAATCAGAA-3’ | ||
| FR1d_t1 | 5’- CAGGAAACAGCTATGACACCTCAGGGTGTCCGAARAAYCARAA-3’ | ||
VERTEBRATE (NON-FISH) PRIMER COCKTAIL For use with mammals, reptiles, amphibians, and some fish and insects Ivanova et. al. (2007). Universal primer cocktails for fish DNA barcoding. Molecular Ecology Notes 7, 544-548 | |||
| LepF1_t1 | 5’- TGTAAAACGACGGCCAGTATTCAACCAATCATAAAGATATTGG-3’ | ||
| VF1_t1 | 5’-TGTAAAACGACGGCCAGTTCTCAACCAACCACAAAGACATTGG-3’ | ||
| VF1d_t1 | 5’-TGTAAAACGACGGCCAGTTCTCAACCAACCACAARGAYATYGG-3’ | ||
| VF1i_t1 | 5’- TGTAAAACGACGGCCAGTTCTCAACCAACCAIAAIGAIATIGG-3’ | ||
| LepR1_t1 | 5’- CAGGAAACAGCTATGACTAAACTTCTGGATGTCCAAAAAATCA-3’ | ||
| VR1d_t1 | 5’- CAGGAAACAGCTATGACTAGACTTCTGGGTGGCCRAARAAYCA-3’ | ||
| VR1_t1 | 5’- CAGGAAACAGCTATGACTAGACTTCTGGGTGGCCAAAGAATCA-3’ | ||
| VR1i_t1 | 5’- CAGGAAACAGCTATGACTAGACTTCTGGGTGICCIAAIAAICA-3’ | ||
INVERTEBRATE PRIMER SET For use with various phyla from the animal kingdom Folmer et al. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology (1994)3(5), 294-299. | |||
| LCO1490 | 5'-TGTAAAACGACGGCCAGTGGTCAACAAATCATAAAGATATTGG-3' | ||
| HC02198 | 5'-CAGGAAACAGCTATGACTAAACTTCAGGGTGACCAAAAAATCA-3' | ||
INVERTEBRATE (ANT) PRIMER COCKTAIL For use with ants Folmer et al. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology (1994)3(5), 294-299. | |||
| LCO1490 | 5'-TGTAAAACGACGGCCAGTGGTCAACAAATCATAAAGATATTGG-3' | ||
| HC02198 | 5'-CAGGAAACAGCTATGACTAAACTTCAGGGTGACCAAAAAATCA-3' | ||
| FormCOId_F | 5’-TGTAAAACGACGGCCAGTATTCAACAAATCAYAAAGAYATYGG-3’ | ||
| FormCOId_R | 5’-CAGGAAACAGCTATGACTAAACTTCIGGRTGWCCAAARAATCA-3’ | ||
FUNGI PRIMER SET For use with fungi Gardes M, Bruns T (1993) ITS primers with enhanced specificity for Basidiomycetes-application to the identification ofmycorrhizae and rusts. Molecular Ecology, 2,113-118. Jackson CJ; Barton RC; Evans EGV (1999). Species identification and strain differentiation of dermatophyte fungi by analysis of ribosomal-DNA intergenic spacer regions. J. Clinical Microbiology, 37, 4: 931-936 | |||
| ITS1F | 5'-TGTAAAACGACGGCCAGTTCCGTAGGTGAACCTGCGG-3' | ||
| ITS4 | 5'-CAGGAAACAGCTATGACTCCTCCGCTTATTGATATGC-3' | ||
FUNGI (LICHEN-SPECIFIC) PRIMER SET For use with lichen Gardes M, Bruns T (1993) ITS primers with enhanced specificity for Basidiomycetes-application to the identification ofmycorrhizae and rusts. Molecular Ecology, 2,113-118. Jackson CJ; Barton RC; Evans EGV (1999). Species identification and strain differentiation of dermatophyte fungi by analysis of ribosomal-DNA intergenic spacer regions. J. Clinical Microbiology, 37, 4: 931-936 | |||
| ITS1F_(Gad) | 5'-TGTAAAACGACGGCCAGTCTTGGTCATTTAGAGGAAGTA-3' | ||
| ITS4> | 5'-CAGGAAACAGCTATGACTCCTCCGCTTATTGATATGC-3' | ||
Primers for Sequencing
5´ TGTAAAACGACGGCCAGT 3´ M13F
5´ CAGGAAACAGCTATGAC 3´ M13R
Messing J., (1983). New M13 vectors for cloning. Methods in Enzymology, 101, 20–78.
Primers mixes designed for use with PCR beads can be ordered from Carolina Biological Supply Company.
Alternatively, individual primers can be ordered from a custom DNA oligonucleotide provider. For instance, Sigma-Aldrich provides an ample supply for many PCR reactions for approximately $15 per primer. We recommend ordering all primers you need at once to save on shipping costs. To order the primers through Sigma:
- Go to the Sigma website, create an account (if needed), and log in.
- On the left side of page, click "products," then under heading Custom Products, click "order oligos and peptides," then "oligos and probes" and finally click "order" under DNA oligos in a tube.
- Enter the oligo name. You can use the primer names indicated above to avoid confusion.
- Select synthesis scale 0.025 µMol
- Select Purification: Desalt
- Paste in the sequence 5' -> 3' listed above with no spaces! As a reminder, these sequences already have the M13 tags included, which will make things easier for you when sequencing.
- Select 5' modification: none
- Select 3' modification: none
- Select Format: dry
Each primer needs to be re-suspended in distilled water upon arrival. We recommend adding enough distilled water to create 100 µM stocks, which can then be diluted to 15 µM for the working solutions to make primer mixes. Stock solutions and working solutions should be stored at -20°C. If at all possible, the tubes should be stored in a non-defrosting freezer, or in a container that keeps tubes frozen during "defrost cycles," as repeated freezing and thawing can degrade the primers.
Ready-To-Go™ PCR beads limit reagent waste and optimize PCR reactions in a classroom setting. Each bead contains reagents so that when brought to a final volume of 25 µL, the reaction contains 2.5 units of Taq DNA polymerase, 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 1.5 mM MgCl2, and 200 µM of each dNTP.
The lyophilized Taq DNA polymerase in the bead becomes active immediately upon addition of the primer/loading dye mix and template DNA. In the absence of thermal cycling, "nonspecific priming" at room temperature allows the polymerase to begin generating erroneous products, which can show up as extra bands in gel analysis. Therefore, work quickly. Be sure the thermal cycler is set and have all experimenters set up their PCR reactions as a coordinated effort. Add primer/loading dye mix to all reaction tubes, then add each student template, and begin thermal cycling as quickly as possible. Hold reactions on ice until all student samples are ready to load into the thermal cycler.
The NEB Taq 2X master mix is a cost-effective alternative to PCR beads and works well in a classroom setting. Taq 2X Master Mix is an optimized ready-to-use solution containing Taq DNA Polymerase, dNTPs, MgCl2, KCI and stabilizers. The Master Mix is used at a 1X final concentration with DNA template and primers in a total reaction volume of 25 µL. It is stable for fifteen freeze-thaw cycles when stored at -20°C or for three months at 4°C, so for frequent use, an aliquot may be kept at 4°C.
1% Cresol Red Dye
Makes 50 mL.
Store at room temperature (indefinitely).
Mix in a 50-mL tube:
- 500 mg cresol red dye
- 50 mL of distilled water
Cresol Red Loading Dye
Makes 50 mL.
Store at –20°C (indefinitely).
- Dissolve 17 g of sucrose in 49 mL of distilled water in a 50-mL tube.
- Add 1 mL of 1% cresol red dye and mix well.
The primer/loading dye mix customizes the PCR reaction for DNA barcoding. The mix incorporates the appropriate primer pair (0.26 picomoles/µL of each primer), 13.8% sucrose, and 0.0081% cresol red. The inclusion of the loading dye components, sucrose and cresol red, allows the amplified product to be directly loaded into an agarose gel for electrophoresis.
Makes enough for 50 reactions. Store at –20°C for 1 year.
Mix in a 1.5-mL tube:
- 640 µL of distilled water
- 460 µL of Cresol Red Loading Dye (see recipes above)
- 20 µL of 15 pmol/µL 5' primer
- 20 µL of 15 pmol/µL 3' primer
(For multiplex primers other than the Invertebrate (Ant) Primer Cocktail, add 20 µL of each primer, and reduce volume of distilled water by 20 µL for each additional primer.)
To mix the Invertebrate (Ant) Primer Cocktail in a 1.5-mL tube:
- 640 µL of distilled water
- 460 µL of Cresol Red Loading Dye (see recipes above)
- 15 µL of 15 pmol/µL LCO1490 5' primer
- 15 µL of 15 pmol/µL HC02198 3' primer
- 5 µL of 15 pmol/µL FormCOId_F 5' primer
- 5 µL of 15 pmol/µL FormCOId_R 3' primer
The primer/loading dye mix customizes the PCR reaction for DNA barcoding. The mix incorporates the appropriate primer pair (0.526 picomoles/µL of each primer), 13.8% sucrose, and 0.0081% cresol red. The inclusion of the loading dye components, sucrose and cresol red, allows the amplified product to be directly loaded into an agarose gel for electrophoresis.
Makes enough for 50 reactions. Store at –20°C for 1 year.
Mix in a 1.5-mL tube:
- 600 µL of distilled water
- 460 µL of Cresol Red Loading Dye (see recipes above)
- 40 µL of 15 pmol/µL 5' primer
- 40 µL of 15 pmol/µL 3' primer
(For multiplex primers other than the Invertebrate (Ant) Primer Cocktail, add 40 µL of each primer, and reduce volume of distilled water by 40 µL for each additional primer.)
To mix the Invertebrate (Ant) Primer Cocktail in a 1.5-mL tube:
- 600 µL of distilled water
- 460 µL of Cresol Red Loading Dye (see recipes above)
- 30 µL of 15 pmol/µL LCO1490 5' primer
- 30 µL of 15 pmol/µL HC02198 3' primer
- 10 µL of 15 pmol/µL FormCOId_F 5' primer
- 10 µL of 15 pmol/µL FormCOId_R 3' primer
- Obtain a PCR tube and use a micropipette with a fresh tip to add 12.5 µL of the master mix to the tube.
- Use a micropipette with a fresh tip to add 10.5 µL of the appropriate primer/loading dye mix (for NEB Taq 2X Master Mix) to each tube.
- Plant cocktail: rbcL primers (rbcLaF / rbcLa rev)
- Fungi cocktail: ITS primers (ITS1F / ITS4)
- Fish cocktail: COI primers (VF2_t1 / FishF2_t1 / FishR2_t1 / FR1d_t1)
- Vertebrate (non-fish): (VF1_t1 / VF1d_t1 / VF1i_t1 / VR1d_t1 / VR1_t1 / VR1i_t1)
- Invertebrate (ant) cocktail: (LCO1490 / HC02198 / FormCOId_F / FormCOId_R)
- Invertebrate cocktail: (LCO1490 / HC02198)
- Place the PCR tubes on ice to prevent premature replication of unwanted primer dimers.
- Use a micropipette with fresh tip to add 2 µL of your DNA (from Part II) directly into the appropriate primer/loading dye mix. Ensure that no DNA remains in the tip after pipetting.
If the reagents become splattered on the wall of the tube, pool them by briefly spinning the sample in a microcentrifuge (with tube adapters) or by sharply tapping the tube bottom on the lab bench.
If your DNA was extracted using Chelex, allow the tubes containing DNA to sit upright for 10 minutes (or centrifuge for 30 seconds) to ensure that any residual Chelex settles on the bottom of the tubes. When removing DNA for PCR, be careful to only pipet from the very top of the liquid to avoid transferring Chelex into the PCR tube as Chelex inhibits PCR.
To use adapters, “nest” the sample tube within sequentially larger tubes: 0.2 mL within 0.5 mL within 1.5 mL. Remove caps from tubes used as adapters.
- Store your sample on ice until your class is ready to begin thermal cycling.
- Place your PCR tube, along with those of the other students, in a thermal cycler that has been programmed with the appropriate PCR protocol.
Amplification from some templates, such as the COI barcode region, may be improved by transferring PCR tubes directly from ice into a hot thermal cycler that has been temporarily paused at the beginning of the first 95°C denaturation step. This limits the formation of undesirable primer dimers. Resume the program when all of the PCR tubes are in the thermal cycler.
Remove caps from 1.5-mL tubes to use as adapters in which to centrifuge 0.5-mL PCR tubes used for PCR amplification. Two adapters are needed to spin 0.2-mL PCR tubes—a capless 0.5-mL PCR tube is nested within a capless 1.5-mL tube.
Amplification of rbcL and COI is simplified by the large number of chloroplast and mitochondrial genomes, which are present at 100-1,000s of copies per cell. ITS is also present at high copy number in most fungi. Thus, the barcode regions are amplified more readily than most nuclear loci and small amount of specimen collected provides enough starting template to produce large quantities of the target sequence, reducing the concentration of contaminants that might inhibit PCR. The recommended amplification times and temperatures will work adequately for most common thermal cyclers, which ramp between temperatures within a single heating/cooling block. IMPORTANT: Follow manufacturer’s instructions for Robocycler or other brands of thermal cyclers that physically move PCR reaction tubes between multiple temperature blocks. These machines have no ramping time between temperatures, and may require longer cycles.
When PCR reactions fail, there are many possible reasons. A common source of difficulty is low quality DNA caused by using too much sample for the isolation. If you suspect your students have used too much material and their PCR failed, consider re-isolating the DNA with the standard protocol while ensuring the students use less material. For dried, degraded, or processed samples, PCR may fail due to low yield, in which case using the appropriate alternative method may allow amplification. For the silica isolation, other possible sources of trouble include evaporation of ethanol from the wash buffer, which can be avoided by storing the wash buffer at low temperature in a sealed container, and failing to remove wash buffer before elution, which can be avoided by carefully removing the wash buffer and drying briefly before elution. Mixing the DNA/silica mixture during incubation may also increase yields.
The PCR may also fail due to changes in the sequence at the primer binding sites, making it impossible to amplify even with high quality DNA. Consulting the literature on the taxa you are studying may help determine whether this is the case. It may also help to re-amplify after lowering the annealing temperature a few degrees, as this may allow the primers to anneal even if the primer binding sites have mutated.
Makes 200 mL.
Use fresh or store solidified agarose for several weeks at room temperature.
- To a 600-mL beaker or Erlenmeyer flask, add 200 mL of 1x TBE electrophoresis buffer and 4 g of agarose (electrophoresis grade).
- Stir to suspend the agarose.
- Dissolve the agarose using one of the following methods:
- Cover the flask with aluminum foil and heat the solution in a boiling water bath (double boiler) or on a hot plate until all of the agarose is dissolved (~10 min).
- Heat the flask uncovered in a microwave oven at high setting until all of the agarose is dissolved (3–5 min per beaker).
- Swirl the solution and check the bottom of the beaker to ensure that all of the agarose has dissolved. (Just before complete dissolution, particles of agarose appear as translucent grains.) Reheat for several minutes if necessary.
- Cover the agarose solution with aluminum foil and hold in a hot water bath (at ~60°C) until ready for use. Remove any “skin” of solidified agarose from the sur- face before pouring the gel.
Notes: Samples of agarose powder can be preweighed and stored in capped test tubes until ready for use. Solidified agarose can be stored at room temperature and then remelted over a boiling water bath (15–20 min) or in a microwave oven (3–5 min per beaker) before use. When remelting, evaporation will cause the agarose concentration to increase; if necessary, compensate by adding a small volume of water. Always loosene cap when remelting agarose in a bottle.
CAUTION: Be sure to electrophorese only 5 µL of each amplified product. The remaining 20 µL must be retained for DNA sequencing: 10 µL for the forward read and, potentially, 10 µL for the reverse read.
Plasmid pBR322 digested with the restriction endonuclease BstNI is an inexpensive marker and produces fragments that are useful as size markers in this experiment. The size of the DNA fragments in the marker are 1,857 bp, 1,058 bp, 929 bp, 383 bp, and 121 bp. Use 20 µL of a 0.075 µg/µL stock solution of this DNA ladder per gel. Other markers or a 100-bp ladder may be substituted.
View and photograph gels as soon as possible after electrophoresis or appropriate staining/destaining. Over time, the small-sized PCR products will diffuse through the gel and the bands they form will lose sharpness.
DNA sequencing of the rbcL, COI or ITS amplicon is required to determine the nucleotide sequence that constitutes the DNA barcode. The forward, the reverse, or both DNA strands of the amplified barcode region may be sequenced. A single, good-quality barcode from the forward strand is sufficient to identify an organism. The majority of database sequences are from the forward strand, so sequencing only the forward strand reduces sequencing cost and simplifies analysis. If you only do a forward read, save the remaining 10 µL of amplicon. If the forward read fails, and time permits, you can send the remainder out to sequence the reverse strand.
However, bi-directional sequencing is important for several reasons. 1) A reverse sequence may provide a readable barcode when the forward sequence fails. 2) Good forward and reverse reads can be combined to produce a consensus sequence that extends the read up to 40 or more nucleotides. This is because the primer itself is not sequenced for either strand, and additional nucleotides downstream from the primer are typically unreadable. Thus, good forward and reverse primers complement these missing sequences, adding most of the primer sequences on either end. 3) One direction may provide a read through a region that is refractory to sequencing in the other direction, such as a homopolymeric region containing a long string of C residues. Thus, the insurance provide by bi-directional sequencing may be worth the added cost, especially if you have need to complete an analysis in a limited time.
Sequencing different barcode regions—rbcL, COI and ITS—and using degenerate and multiplex primers complicate DNA sequencing. Strictly speaking, each different primer would need to be provided for forward and reverse sequencing reactions. As a work-around for this problem, the primers used in this experiment incorporate a universal M13 primer sequence. In addition to a sequence specific to the rbcL, COI or ITS barcode locus, the 5’ end of each primer has an identical 17 or 18 nucleotide sequence from the bacteriophage vector M13.
In the traditional approach to genome sequencing, genomic DNA is cloned into an M13 vector. Then a universal M13 primer is used to sequence the genomic insert just downstream from the primer. This same strategy is used in sequencing rbcL, COI and ITS barcodes in this experiment. During the first cycle of PCR, the M13 portion of the primer does not bind to the template DNA. However, the entire primer sequence is covalently linked to the newly-synthesized DNA and is amplified in subsequent rounds of PCR. Thus, the M13 sequence is included in every full-length PCR product. This allows a sequencing center to use universal forward and reverse M13 primers for the PCR-based reactions that prepare any rbcL, COI or ITS amplicon for sequencing.
The sequence of the M13 forward and reverse primers are:
- M13F: TGTAAAACGACGGCCAGT
- M13R: CAGGAAACAGCTATGAC
We recommend using GENEWIZ for DNA barcode sequencing. GENEWIZ has optimized reaction conditions for producing the barcode sequences in this laboratory and produces excellent quality sequence with rapid turnaround—usually within 48 hours of receipt of samples. GENEWIZ sequences are automatically uploaded to the DNALC’s DNA Subway 2.0 website. Before submitting samples for sequencing, consult the GENEWIZ guide.
Getting Started: GENEWIZ DNA Sequencing Services
- Go to www.GENEWIZ.com and click “LOGIN,” then select "Sign up now" to create a user account.
- You will receive an Activation Email at the email address used as the Username from Genewiz, and you will need to verify the email address by clicking the activation link provided in the message.
- After signing into your user account, update your Profile, found under the My Account heading.
- When creating your profile enter your institution name followed by “-DNALC” (very important!) , then your personal information so invoices are sent to the correct location.
- When prompted to select the “Methodologies Used”, select “Sanger Sequencing”.
- Obtain a valid Purchase Order number from your Purchasing department or use a valid credit card. Contact GENEWIZ for pricing information.
Preparing Your PCR Product
- If you have not sequenced samples at GENEWIZ before, consult their detailed guide.
- Verify that you have obtained PCR product of the correct length and with visible concentration on an agarose gel.
- Prepare 8-strip 0.2 mL PCR tubes appropriate for the number of samples you wish to submit. If you will be submitting a large number of samples (≥48), you can submit up to 94 PCR products per 96 well plate.
- You will need to have 10 µL of PCR product for each sequencing reaction. Samples do not need to be split into two tubes to receive forward and reverse sequencing. For example: if you have 8 samples that need forward and reverse sequencing (16 reactions in the chart), you can send the samples in 8 tubes as long as you list all forward reads first, followed by all reverse reads.
Submitting a Sample for Sequencing
- Log in to your user account to place your sequencing order.
- Select “SANGER SEQUENCING” from the list.
- Select “Comprehensive Order,” and a list will appear.
- From that list, select “PCR Product – Un-Purified.”
- Select “Custom for the Service Type.”
- For sequencing priority, select “Standard.”
- For DNA concentration, it is best to send in a gel image with representative samples alongside a marker. This will be used by GENEWIZ to calculate the correct amount of clean-up reagents to use and the amount of product to use in the sequencing reaction. If a gel image is not supplied, GENEWIZ will use our default amount for setting up the sequencing reactions (5 ng/µL).
- You can choose either the online form or upload excel form. If you choose to use the online form, you will need to enter the number of samples.
Filling Out the Sample Form
- Enter a sample name for each sample. This could be a number or initials, etc.
- For “DNA Length (vector + insert in bp),” enter 501-1000 for rbcL, COI, and ITS.
- Leave the My Primer section empty. For Genewiz Primer: select “M13F” to sequence the forward strand, and “M13R” to sequence the reverse strand.
To create a consensus barcode sequence, each sample should be sequenced in the forward and reverse direction. To do so, you will need to enter all of the sample information for the reverse reads after all sample information for the forward reads. Example table: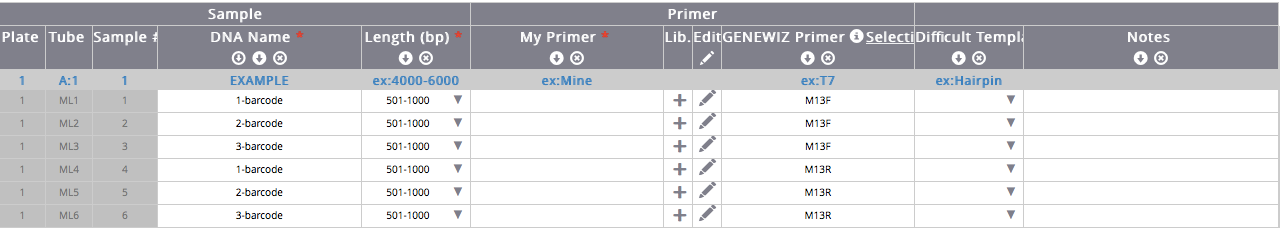
- Click “Save & Review.”
- Carefully review your form, then click “Add to Cart.”
- Enter your payment information, and click “Check Out.”
- Review your order, then click “Submit.”
Shipping Samples to GENEWIZ
- Print a copy of the order form, and mail it along with your samples.
- Be sure that the tubes are labeled exactly the same in the gel photo and on the order form. Failure to do so may delay sequencing or make it impossible to complete. Email DNALCSeq@cshl.edu if you need help.
- Ship your samples via standard overnight delivery service (Federal Express, if possible).
- Pack your samples in a letter pack or small shipping box, padding samples to prevent too much shifting. Room temperature shipping—with no ice or ice pack—is expected. PCR products are stable at ambient temperature, even if shipped on a Friday for Monday delivery.
- Address the shipment to GENEWIZ at the following location:
GENEWIZ, Inc.
115 Corporate Blvd.
South Plainfield, NJ 07080
Additional GENEWIZ shipping options:
In some cases, based on your location, you may be able to reduce shipping costs by using a GENEWIZ drop box. Call 1-877-436-3949 to find out if one is available in your area.
